
Ruta metabòlica del grup hemo
|
|
S'ha proposat fusionar aquest article a «Grup hemo». (Vegeu la discussió, pendent de concretar). Data: 2024 Motiu: context |
La ruta metabòlica del grup hemo, també anomenada Shemin pathway, és un conjunt de reaccions químiques que es donen per a la síntesi i degradació del grup hemo, una porfirina sintetitzada, principalment, a la medul·la òssia i al fetge.[1]

La biosíntesi d'aquest grup de molècules resulta essencial per a la formació d'hemoglobina, una proteïna multifuncional que transporta oxigen des dels pulmons fins a múltiples teixits de l'organisme.[2] Altrament, a la ruta anabòlica, la disponibilitat i correcte tràfic intercel·lular i intracel·lular del ferro són indispensables, ja que el darrer pas implica la inserció d'ions ferrosos, pel fet que l'hemo és primordial pel seu emmagatzematge i reducció.[3]
La rellevància de la degradació d'aquest grup porfíric resideix en el fet que és una resposta conservada evolutivament per contrarestar l'estrès oxidatiu, atès que sota aquestes condicions el grup hemo és alliberat de l'hemoglobina. Consegüentment, sense l'existència d'aquesta via catabòlica, es produiria una acumulació d'aquest grup de molècules a l'organisme, les quals esdevindrien tòxiques perquè catalitzarien radicals lliures, que són altament reactius, i, per tant, podrien reaccionar amb altres biomolècules.[4]
La presència de mutacions que afecten enzims involucrats en la síntesi del grup hemo donen lloc a una sèrie de patologies, com és el cas de porfíria i anèmia. Tanmateix, una deficiència a les rutes catabòliques provoca hiperbilirubinèmia.[5]
Anabolisme
[modifica]La síntesi del grup hemo consta de 8 processos anabòlics que tenen lloc principalment a la medul·la òssia i al fetge. L'hemo que se sintetitza a la medul·la òssia correspon a un 80% del total produït a l'organisme i s'utilitza majoritàriament per a la formació d'hemoglobina. En canvi, el que se sintetitza al fetge, que correspon a un 15%, s'usa principalment per a la síntesi de citocroms.[6] Aproximadament, se sintetitzen entre 40 i 50 mil·ligrams d'hemo diàriament. [2]
La rellevància de la biosíntesi del grup hemo resideix en el fet que forma part d'un gran nombre de molècules com són el citocrom (P450), la mioglobina, encarregada de l'emmagatzemament d'oxigen al teixit muscular, la peroxidasa, que catalitza la degradació de peròxid d'hidrogen, o l'hemoglobina, la qual transporta oxigen dels pulmons a múltiples teixits.[7]
La biosíntesi s'inicia als mitocondris, posteriorment és traslladada al citosol i, darrerament, finalitza de nou als mitocondris. [3]
L'ALA-sintasa (ALAS) és l'enzim regulador de la síntesi de l'hemo al fetge i a la medul·la òssia. Dos gens situats en dos cromosomes diferents en els humans codifiquen les dues formes ALAS de l'enzim: l'ALAS I, de naturalesa hepàtica, i l'ALAS II, de naturalesa eritroide (medul·la òssia). Totes les cèl·lules expressen l'ALAS I mentre que les de la medul·la òssia només expressen l'ALAS II.[7] Els esdeveniments post-transcripcionals i els mecanismes reguladors per a l'expressió dels dos gens ALAS són diferents per diferenciar els eritròcits dels de la resta de cèl·lules. Per exemple, l'ALAS I controla la producció d'hemo per a les funcions cel·lulars bàsiques.[7]

En primer lloc, se sintetitza l'àcid 5-aminolevulínic (ALA) en la matriu mitocondrial. Aquest procés és catalitzat per l'ALAS, que limita la velocitat de la síntesi de l'hemo, regulant la condensació de la glicina i succinil CoA. Per portar-ho a terme es requereix una molècula de piroxidat fosfat (PLP o vitamina B6) que actuï com a cofactor. En aquesta reacció s'allibera CoA, CO₂ i protons.[1][8][7]
Posteriorment, es produeix la formació de porfobilinogen (PBG), pel qual és necessari que l'ALA formada surti dels mitocondris i sigui exportada al citosol. Aquest canvi de localització el duu a terme la proteïna SLC25A38. Ja al citosol, dues molècules d'ALA es deshidraten gràcies a l’enzim ALA deshidratasa, que requereix zinc. Com a resultat es forma PBG i s'eliminen dues molècules d'aigua i protons. El PBG té una estructura heterocíclica formada per quatre carbonis i un nitrogen (anell pirròlic). Endemés, conté dues cadenes laterals, un radical acetat i un radical propionat. La seva formació es veu afectada per la intoxicació de plom, ja que l'ALA deshidratasa és inhibit per aquest i l'acumulació de l'enzim pot derivar a problemes neuronals. [4][Enllaç no actiu]
A continuació, al citosol, l'enzim PBG desaminasa s’uneix al PBG alliberant una molècula d'amoni (NH4). A aquesta molècula se li uneix un altre PBG, successivament fins que s'uneixin 4 molècules (amb 4 anells pirrols, formant un tetrapirrol lineal). Aquest nou compost format s'anomena hidroximetilbilà lineal. Seguidament, una molècula d'aigua provoca un tall que causa que aquesta molècula se separi de l'enzim i quedi lliure, permetent la seva ciclació formant un anell de pirrol asimètric D. Aquest pas és crític, ja que una formació incorrecta d'anell de porfirina condueix a protoporfíria.[5]
Depenent de si s'empra un enzim o no, aquesta molècula pot tenir dos destins: l'uroporfirinogen III, catalitzat per l'uroporfirinogen-III sintasa (present en un 80% dels casos) o uroporfirinogen I, el qual no necessita cap enzim, és a dir, es formaria per un plegament espontani (en un 20% dels casos). Així doncs, l'uroporfirinogen I serà eliminat per l’orina i, si hi trobem oxigen, s'oxidarà i formarà la uroporfirina. En canvi, l'uroporfirinogen III seguirà la ruta metabòlica.[9]
Aquesta molècula d'uroporfirinogen III es descarboxila formant coproporfirinogen III (CPG III), gràcies a l'enzim uroporfirinogen III descarboxilasa, i s'alliberen 4 molècules de diòxid de carboni.[5][Enllaç no actiu] El CPG III es pot eliminar per la femta, donant-li el seu color característic marró, o seguir la ruta metabòlica. En el darrer cas, el CPG III torna al mitocondri i s'oxida, generant protoporfirinogen III mitjançant l'enzim coproporfirinogen III oxidasa. [6] Posteriorment, pateix una última oxidació, regulada per l'enzim protoporfirinogen III oxidasa, que forma la molècula de protoporfirina IX.
La reacció final implica la inserció de ferro fèrric (Fe2+) a la protoporfirina IX, catalitzada per l'enzim ferroquelatasa, que condueix a la formació d'hemo.[3]
Síntesi de l'hemo a les cèl·lules eritroides i hepàtiques
[modifica]Els les cèlules eritroides de la mèdula ossia i els hepatòcits presenten un predomini de la síntesi d'aquest grup de molècules, fet que reflecteix una major demanda del grup hemo. Aquest succés es deu al fet que aquests dos tipus de cèl·lules són encarregades de la síntesi de molècules que presenten aquest grup, com és el cas de l'hemoglobina i el citocrom P450.[7]
La síntesi del grup hemo amb el propòsit de ser incorporat en l'hemoglobina té repercussió en el fet que els eritròcits immadurs són estimulats per aquest grup, perquè condueix a una major síntesi de cadenes de globina i eritropoetina, necessàries en la maduració dels eritroblasts. Quan els eritroblasts maduren, s'atura la síntesi del grup hemo i la d'hemoglobina. Endemés, la biosíntesi de l'hemo als eritròcits està controlada per la disponibilitat de ferro intracel·lular, el qual forma part de l'estructura de l'hemoglobina.[3]
D'altra banda, la síntesi de l'hemo al fetge varia considerablement i està extremadament regulada, ja que aquest grup ubicat fora de les proteïnes en altes concentracions provoca danys als hepatòcits. La síntesi d'hemo s'atura quan aquest no és incorporat a les proteïnes i quan aquest grup i l'hemina (nom farmacològic de l'hemo) s'acumulen. L'hemina disminueix la síntesi de l'ALAS I de diferents maneres: redueix la síntesi desestabilitzant l'ARNm que codifica per la formació d'ALAS I o inhibeix la importació d'aquesta del citosol al mitocondri.[9]
Metabolisme del ferro
[modifica]Degut al fet que la biosíntesi de l'hemo requereix ferro, es tracta d'una ruta anabòlica que depèn de la seva disponibilitat a escala intracel·lular.
Les principals fonts de ferro són la ingesta d'aliments, la descomposició de productes que el contenen, com és el cas de l'hemoglobina, i l'alliberament de les reserves reticuloendotelials.[3]

Les cèl·lules humanes l'absorbeixen mitjançant enteròcits del duodè i el jejú proximal, a través de l'endocitosi regulada per receptors de transferrina de complexos circulants de Fe³⁺. Seguidament, el ferro en estat fèrric (Fe³⁺) unit a la transferrina és reduït a estat ferrós (Fe²⁺), a causa de l'acció de la metalloreductasa STEAP3 que es troba a la superfície dels enteròcits. Posteriorment, el transportador de metalls divalents (DMT1) transporta Fe²⁺ des de la superfície apical dels enteròcits a l'interior de la cèl·lula. D'aquesta manera, entra a l'espai intermembranós del mitocondri, on és transportat a través de la membrana interna i cap a la matriu per la Mitoferrina1 (MFRN1, SLC25A37) en eritroblasts i la Mitoferrina2 (MFRN2, SLC25A28) en cèl·lules no eritroides. Altres possibles transportadors són els endosomes i el transportador del grup hemo.[7]
Finalment, el Fe²⁺ s'oxida a Fe³⁺ amb l'ajuda de l'hefaestina i passa a la circulació unit a la transferrina, de manera que el ferro és transportat als teixits. La transferrina, que per hematopoesi transporta ferro a la medul·la òssia, al fetge i altres òrgans per al seu emmagatzematge, s'uneix als receptors de transferrina, que s'expressen de forma significativa en cèl·lules amb gran demanda de ferro. Endemés, aquesta proteína transporta ferro pels processos cel·lulars que en requereixen.
Catabolisme
[modifica]La ruta catabòlica del grup hemo consisteix en la seva descomposició. Els components resultants poden ser reutilitzats per la síntesi de nous grups hemo o eliminats de l'organisme. Des d'una perspectiva general, resulta a la producció de pigments biliars i bilirubina, que, posteriorment, és excretada a través de la bilis.[10]
El catabolisme del grup hemo juga un paper fonamental en la protecció cel·lular, l'apoptosi i la inflamació, entre altres processos fisiològics i patològics.
La degradació del grup hemo és iniciada en els macròfags, ubicats en la melsa, els quals suprimeixen de la circulació sanguínia tots aquells eritròcits que presenten alteracions que comporten conseqüències negatives. Aquestes cèl·lules sanguínies són degradades pel sistema reticuloendotelial i són capaces d'alliberar hemoglobina quan es produeix el seu trencament.[10]

A escala bioquímica, s'oxida el grup hemo, per l'acció de l'enzim hemooxigenasa, i en alliberar protons, l'hemo esdevé biliverdina. Endemés, es produeix monòxid de carboni (CO), que és eliminat pels pulmons, i Fe2+, el qual pot ser reutilitzat en la síntesi d'hemoglobina.[11]
La biliverdina és transformada mitjançant una reacció de reducció a bilirubina, amb la intervenció de la biliverdina reductasa. Aquesta bilirubina indirecta actua com a substància tòxica per a l'organisme. De manera immediata, l'albúmina present en la sang capta dues molècules de bilirubina indirecta. Consegüentment, la primera és fermament unida, però la segona molècula presenta una unió làbil, motiu pel qual manifesta l'habilitat de desprendre's de la unió i, per tant, és capaç de penetrar sense gran complexitat el teixit nerviós.[10]
Tanmateix, la bilirubina conjugada (primera molècula captada per l'albúmina) és transportada a l'intestí prim a través del conducte biliar i, seguidament, a l'intestí gros. De manera consecutiva, a causa de l'acció bacteriana, la bilirubina directa és reduïda a urobilinogen. Una petita fracció és reabsorbida i s'excreta per via renal, fet que és responsable del color groc a l'orina. Aquest urobilinogen ubicat a l'intestí gros esdevé estercobilinogen, el qual pateix un procés oxidatiu, convertint-se en estercobilina, responsable del color marró de la femta.[10]
Finalment, l'urobilinogen de l'intestí es reabsorbeix per circulació enterohepàtica i retorna al fetge per la sang portal.[11]
Patologies associades
[modifica]Qualsevol alteració de les reaccions químiques en el metabolisme del grup hemo presenta associada una patologia, com per exemple la porfíria i l'anèmia, associades a l'anabolisme, i la hiperbilirubinèmia neonatal, associada al catabolisme.
Porfíria
[modifica]La porfíria és un grup de trastorns poc freqüents originats per l'acumulació de porfirines, les quals són necessàries per a la síntesi de l'hemo.[12] Aquest grup de patologies és causat per una deficiència en l'activitat d'enzims que intervenen en les rutes anabòliques del grup hemo, fet que provoca una considerable acumulació de metabòlits intermediaris. Consegüentment, es generen una sèrie d'afeccions, principalment, al sistema nerviós i a la pell.[13]
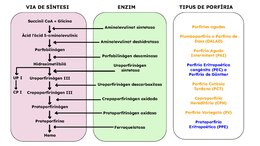
Segons quin enzim involucrat en la biosíntesi d'aquest grup de molècules presenta la deficiència, es distingeixen 7 categories diferents de porfíries. Tanmateix, de manera general, d'acord amb el teixit en què predomina el defecte, les porfíries són subdividides en hepàtiques i eritropoètiques.[14] Segons la simptomatologia que presenten, es poden dividir en agudes, cutànies i mixtes.
Les porfíries agudes, les quals es troben en la categoria de les hepàtiques, són les més freqüents. Aquesta patologia presenta el seu origen en la deficiència de l'enzim ALAS, fet responsable de l'excés de precursors de molècules ALA, les quals actuen com a neurotoxines. Aquestes afecten el sistema nerviós i causen dolor abdominal i paràlisi muscular, que pot afectar les quatre extremitats.[13][15]
Altrament, hi ha organismes que també desenvolupen afeccions cutànies originades per alteracions en l'ALAS. Consegüentment, s'originen múltiples cremades i cicatrius significants a causa de la foto exposició de zones amb elevada concentració de porfirines.[13][16][17]
Encara que a la major part dels casos les porfíries són heretades per factors genètics, la porfíria cutània tardana (PCT) és l'única que també pot ser produïda per factors ambientals, els quals causen una reducció significativa de l'activitat de l'uroporfirinogen descarboxilasa, ubicat en el fetge.[17][18]
Anèmia
[modifica]Malgrat que no s'acostuma a associar l'anèmia amb la presència d'anomalies concretes de la ruta metabòlica del grup hemo, aquesta patologia pot ser originada per una deficiència de la piridoxina[19] a causa de la implicació del fosfat de piridoxal, la forma activa de la vitamina B6, a la síntesi de l'àcid aminolevulínic durant l'anabolisme de l'hemo. Com aquesta vitamina participa en la síntesi d'hemoglobina, el transport d'oxigen a la sang no és adequat, fet característic de l'anèmia.[20][21]
No obstant això, també es presenten anèmies sideroblàstiques un grup heterogeni de trastorns que es deuen a l'emprament defectuós del ferro. Així doncs, independentment del fet que la deficiència sigui adquirida o congènita, l'organisme presenta una incapacitat d'incorporar l'element a la protoporfirina IX, fet que comporta la formació de sideroblasts a l'anell.[22] Tanmateix, l'origen de l'ús incorrecte resideix en la inadequada utilització medul·lar del ferro, malgrat que les concentracions d'aquest siguin correctes.[23]
Hiperbilirubinèmia neonatal
[modifica]La hiperbilirubinèmia neonatal, també anomenada icterícia, és una patologia associada a deficiències en les reaccions catabòliques del grup hemo que causen, majoritàriament, una producció excessiva de bilirubina, fet responsable de la característica coloració de tonalitat groga tant en pell com ulls.
Segons quina sigui l'anomalia en el catabolisme, la icterícia pot ser classificada en tres grans categories.
En primer lloc, la icterícia pot ser originada per un increment en la producció de bilirubina, ja que els nadons presenten una nombrosa quantitat d'eritròcits estructuralment deficients, fet que indueix un major alliberament d'hemoglobina i, per consegüent, una hiperactivació de ruta catabòlica del grup hemo.[10]
Endemés, considerables casos d'icterícia són originats per una disminució en la captació i conjugació hepàtica, afavorint el fet que una abundant quantitat de bilirubina penetri en el teixit nerviós, perquè aquesta queda alliberada de la unió amb l'albúmina. Aquest vessant d'icterícies resulta la més greu; en el cas de derivar en una encefalopatia, el pacient podria presentar, fins i tot, paràlisi cerebral i complexos dèficits sensoriomotors.[24]
Finalment, l'excedent de bilirubina també pot ser desencadenat per una dificultat en la seva reducció per l'acció bacteriana, atès que l'organisme presenta una sèrie d'impediments per arribar fins a l'intestí.[10]
Bibliografia
[modifica]- ↑ 1,0 1,1 Chen, Caiyong; Chung, Jacky; Paw, Barry H. «Heme Metabolism and Erythropoiesis». Current Opinion in Hematology, 2012, 19(3), pàg. 156-162.
- ↑ Peñuela, Oscar Andrés «Hemoglobina: una molécula modelo para el investigador». Colombia Médica, Setembre 2005, 36(3), pàg. 215-225.
- ↑ 3,0 3,1 3,2 3,3 Boccio, José; Salgueiro, Jimena; Lysionek, Alexis; Zubillaga, Marcela; Goldman, Cinthia, et al. «Metabolismo del hierro: conceptos actuales sobre un micronutriente esencial». Archivos latinoamericanos de nutrición, Juny 2003, 53(2), pàg. 119-132.
- ↑ Heeba, Gehan H.; Mahmoud, Magda E.; Hanafy, Amr «Anti-inflammatory potential of curcumin and quercetin in rats: Role of oxidative stress, heme oxygenase-1 and TNF-α». Toxicology and industrial health, 2014, 30(6), pàgs 551-560.
- ↑ 5,0 5,1 Fujiwara, Tohru; Harigae, Hideo «Biology of Heme in Mammalian Erythroid Cells and Related Disorders». BioMed Research International, Octubre 2015, 2013, pàg. 61-65.
- ↑ Kloehn, Joachim; Harding, Clare R.; Soldati-Favre, Dominique «Supply and demand—heme synthesis, salvage and utilization by Apicomplexa». The FEBS journal, 2021, 288(2), pàg. 382-404.
- ↑ 7,0 7,1 7,2 7,3 7,4 7,5 Kumari, Asha. Sweet Biochemistry: Remembering Structures, Cycles, and Pathways by Mnemonics. Octubre 2017. Academic Press, p. 33-36.
- ↑ Casas, Ariadna «Biosíntesis del hemo». Metabolismo del hemo y neoplasias, 1996, 1, pàg. 1-12.
- ↑ 9,0 9,1 To-Figueras, Jordi «Biosynthesis of heme and the porphyrias». Elsevier Journal, Septembre 2023, 159(1), pàg. 51-57.
- ↑ 10,0 10,1 10,2 10,3 10,4 10,5 Mazzi Gonzales de Prada, Eduardo «Hiperbilirrubinemia neonatal». Revista de la Sociedad Boliviana de Pediatría, Gener 2005, 44(1), pàg. 26-35.
- ↑ 11,0 11,1 Bauer, Michael «Heme oxygenase in liver transplantation: heme catabolism and metabolites in the search of function». Journal of Hepatology, Agost 2003, 38(2), pàg. 286-288.
- ↑ Sood, Siddharth; Mingos, Nicholas; Ross, Gayle «Porphyria». The New England journal of medicine, 2017, 377(21), pàg. 2100-2101.
- ↑ 13,0 13,1 13,2 Rossetti, María Victoria; Buzaleh, Ana María; Fukuda, Haydée; Lombardo, María Elisa et al. «Metabolismo del hemo: las dos caras de los efectos de la acumulación de precursores y porfirinas». Acta bioquímica clínica latinoamericana, 50(4), pàg. 547-573.
- ↑ «Tipos de porfirias». Asociación española de porfiria, 2023.
- ↑ «Porfiria hepática aguda: causas, síntomas y tratamientos». American Liver Foundation, 01-05-2023.
- ↑ «Porfíries i síndrome de Rett». Redacció del 324Cat, 01-07-2020.
- ↑ 17,0 17,1 Philipps, John D. «Heme biosynthesis and the porphyrias». Molecular Genetics and Metabolism, Novembre 2019, 128(3), pàg. 164-177.
- ↑ Dailey, Harry A.; Meissner, Peter N. «Erythroid heme biosynthesis and its disorders». Cold Spring Harbor Perspectives in Medicine, Abril 2013, 3(4), pàg. 10-11.
- ↑ Johnson, Larry E. «Deficiencia y dependencia de Vitamina B6», 01-11-2022.
- ↑ Giménez Serrano, Salvador «Anemias». Formación Médica Continuada en Atención Primaria, Maig 2004, 18(5), pàg 62-69.
- ↑ Spinekker, A.; Sola, R.; Lemmen, V.; Castillo, M.J.; Pietrzik, K., et al «Vitamin B6 status, deficiency and its consequences - an overview». Nutrición Hospitalaria, Gener 2007, 22(1), pàg. 7-24.
- ↑ Martínez- Sánchez, Lina María; Castañeda, Santiago «Anemia sideroblástica una enfermedad infrecuente de causas múltiples». Revista Cubana de Hematología, Inmunología y Hemoterapia, Febrer 2021, 36(3), pàg. 1-14.
- ↑ Fujiwara, Tohru «Sideroblastic Anemia». Rinsho Ketsueki, 2019, 60(5), pàg. 408-416.
- ↑ Campistol, J.; Galvez, H.; García Cazorla, A.; Málaga, I.; Iriondo, M., et al «Disfunción neurológica inducida por bilirrubina». Elsevier Journal, Març 2010, 27(4), pàg. 202-211.